
CagriSema Research: Combining Cagrilintide and Semaglutide Dosing
CagriSema is Novo Nordisk’s investigational fixed-dose combination of cagrilintide 2.4

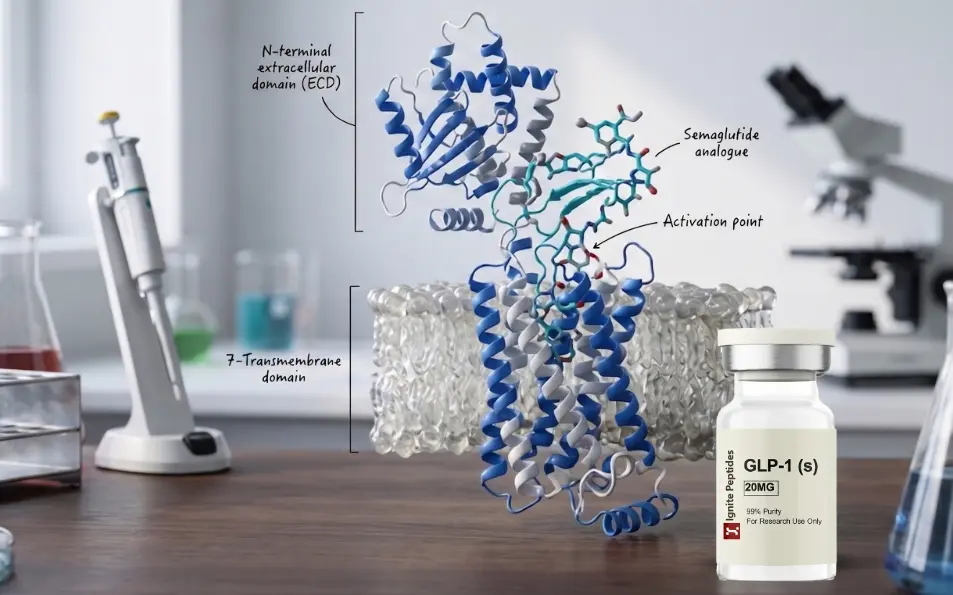
Research on the GLP-1 mechanism is one of the fastest-growing areas in molecular biology. GLP-1 stands for glucagon-like peptide-1. It is an incretin hormone. The body produces it in the gut and uses it to regulate a wide range of cellular processes.
The GLP-1 mechanism begins when the hormone binds its receptor, GLP-1R. Classified within the class B subfamily, this receptor operates as a G protein-coupled receptor. Once activated, it starts a chain of molecular events inside cells. These events include a sharp rise in cyclic AMP, activation of two key protein kinases, and calcium-driven cellular responses.
Semaglutide is a GLP-1 receptor agonist. It copies the GLP-1 mechanism. But it was engineered to resist rapid breakdown and stay active for much longer than native GLP-1.
This article explains the full GLP-1 signalling pathway. It covers biosynthesis, receptor structure, the intracellular signalling cascade, semaglutide pharmacology, tissue distribution, and biased agonism. All content is drawn from peer-reviewed research published in Nature, PubMed/PMC, Frontiers in Endocrinology, and PNAS.
Researchers sourcing lab-verified GLP-1 research peptides can explore Ignite Peptides’ catalogue of >99% purity, COA-backed compounds.
| The GLP-1 mechanism activates a class B GPCR, raises intracellular cAMP via adenylyl cyclase, and triggers a PKA-EPAC2 signalling cascade that closes KATP channels, depolarises the cell membrane, and drives calcium-dependent cellular responses. For research dosing reference, see the GLP-1 (S) Dosage Calculator. |
GLP-1 originates from the preproglucagon gene. This gene encodes a large protein called proglucagon. In intestinal L-cells and neuronal tissue, the enzyme prohormone convertase PCSK1/3 cleaves proglucagon. This produces two active forms: GLP-1(7-36) amide and GLP-1(7-37).
The same proglucagon gene is expressed in pancreatic alpha-cells. But there, a different enzyme, PCSK2, processes it instead. PCSK2 produces glucagon rather than GLP-1. This is called tissue-specific processing. The precursor is the same. The enzyme is different. The product is different.
L-cells release GLP-1 in response to nutrients. Fatty acids, glucose, and bile acids all trigger secretion. After release, GLP-1 enters portal circulation and begins travelling toward target tissues.
The GLP-1 mechanism faces a major obstacle after secretion. The enzyme dipeptidyl peptidase-4 (DPP-4) degrades GLP-1 almost immediately. DPP-4 cleaves the bond between Ala8 and Glu9 at the N-terminus. This produces GLP-1(9-36), which is the inactive metabolite.
DPP-4 is expressed on intestinal endothelial cells. These cells sit right next to L-cell secretion sites. So degradation begins before GLP-1 even leaves the gut. Research shows that less than 25% of secreted GLP-1 exits the gut intact.
In the liver, 40-50% of the remaining active GLP-1 is further degraded. The overall result: only 10-15% of secreted GLP-1 reaches systemic circulation in active form.
Native GLP-1 has a plasma half-life of just 1-2 minutes. This rapid inactivation is the central pharmacokinetic challenge that led to the development of DPP-4-resistant analogues like semaglutide.
The glucagon-like peptide-1 receptor (GLP-1R) falls under the class B category of GPCRs. It has a seven-transmembrane (TM) domain topology. This architecture is shared by all GPCRs. But class B GPCRs have an extra feature: a large N-terminal extracellular domain (ECD). The ECD is the primary site for peptide recognition.
Class B GPCRs bind long polypeptide hormones. GLP-1, glucagon, GIP, PTH, and related peptides all target class B receptors. Cryo-EM structural studies have resolved the activated GLP-1R-Gs complex at near-atomic resolution. These studies confirm the precise binding geometry and activation mechanism of the receptor.
GLP-1 binds its receptor through a two-domain model. This process has four sequential steps:
Step 1: C-terminal docking: The C-terminal alpha-helix of GLP-1 docks onto the receptor ECD. This is the initial contact point.
Step 2: N-terminal insertion: The N-terminus of GLP-1 inserts into the transmembrane bundle core. This is the activation-triggering step.
Step 3: Loop stabilisation: Extracellular loops of the receptor clamp around the bound peptide. This stabilises the active conformation.
Step 4: TM6 pivot: Transmembrane helix 6 (TM6) bends sharply at its midpoint. Its intracellular half rotates outward. This creates space for the G protein heterotrimer alpha-5 helix and starts intracellular signalling.
For semaglutide, specific residue interactions strengthen receptor binding. Hydrogen bonds between Val33 and receptor residue Arg121, and ionic bonds between Arg36 and Glu68, provide more polar contacts than native GLP-1. These extra interactions explain semaglutide’s longer receptor residency time.
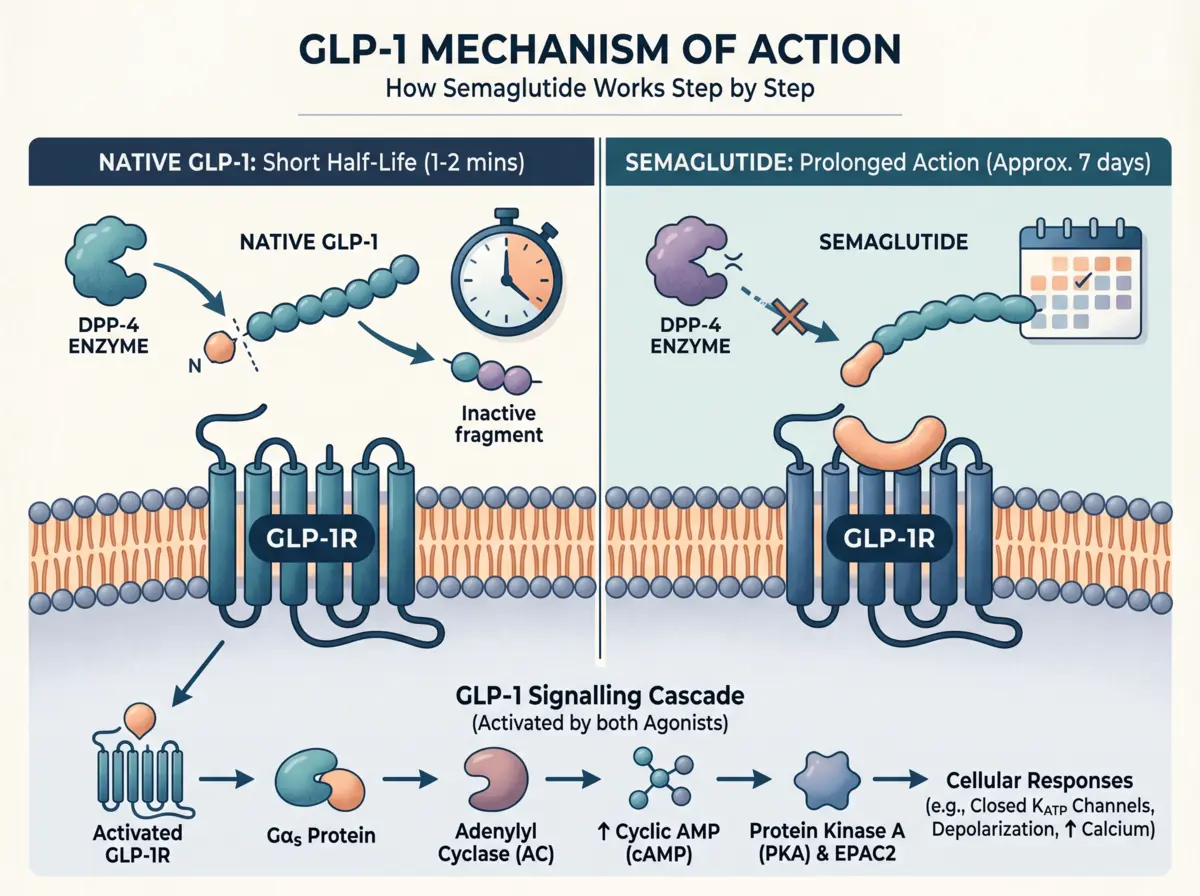
This section is the core of the GLP-1 mechanism. The signalling cascade moves from receptor activation to a cellular response. Each step depends on the one before it.
GLP-1 binding activates the Gαs subunit of the trimeric G-protein complex. Gαs dissociates from the receptor-G-protein complex. It then activates plasma membrane-bound adenylyl cyclase (AC).
AC converts ATP into cyclic AMP (cAMP). Intracellular cAMP levels rise within seconds of receptor activation. This rapid cAMP rise is the starting signal for everything downstream.
| Research Note: In murine pancreatic beta-cells, GLP-1R activates both Gαs and Gαq. The GIP receptor activates only Gαs. This difference in G protein coupling is a key distinction between the two incretin hormone receptors. (Frontiers in Endocrinology, 2024) |
Elevated cAMP activates two effector proteins. The first is protein kinase A (PKA). The second is exchange protein activated by cAMP-2 (EPAC2). Both are part of the core GLP-1 mechanism.
PKA phosphorylates multiple downstream proteins. It activates the transcription factor CREB (cAMP response element-binding protein). CREB promotes insulin gene expression. PKA also directly phosphorylates components of the insulin secretion machinery.
EPAC2 works differently. It lowers the ATP concentration required to close KATP channels. As a result, the threshold required to trigger membrane depolarisation is lowered. PKA and EPAC2 act together; both are needed for a full cellular response.
KATP channels are potassium channels. They consist of Kir6.2 and SUR1 subunits. Kir6.2 acts as a glucose/ATP sensor. Under low-glucose conditions, the channel stays open. Potassium ions flow out. The membrane holds a resting potential of -65 to -53 mV.
When glucose levels rise, intracellular ATP increases. ATP causes KATP channels to close. GLP-1 signalling through EPAC2 and PKA enhances this closure. With the channel closed, K+ accumulates inside the cell. The membrane depolarises.
This step is glucose-dependent. The GLP-1 mechanism does not cause full membrane depolarisation without sufficient glucose-derived ATP. This glucose dependence is a fundamental feature of the signalling pathway.
Membrane depolarisation opens voltage-dependent calcium channels (VDCCs). Ca2+ enters the cell from the extracellular space. This calcium influx then triggers calcium-induced calcium release (CICR) from the endoplasmic reticulum. Cytosolic Ca2+ rises sharply.
Elevated Ca2+ drives two sequential events. First, secretory granules move to the cell membrane, which is called priming. Granules merge with the cell membrane and discharge their contents. This process is called exocytosis. The full sequence from GLP-1R activation to secretion takes minutes under glucose-sufficient conditions.
A major new finding in GLP-1 mechanism research: cAMP generation continues inside endosomes after receptor internalisation.
When GLP-1 binds GLP-1R, the receptor-ligand complex is eventually internalised into endosomes. Earlier research assumed GPCR signalling stopped after internalisation. New studies show otherwise. Inside endosomes, the GLP-1R-ligand complex remains active. It continues producing cAMP via endosomal adenylyl cyclase.
Blocking endocytosis reduces overall GLP-1R-mediated signalling output. This confirms that endosomal cAMP is a functional part of the GLP-1 mechanism, not just a recycling event. This finding was published in the American Journal of Physiology-Endocrinology and Metabolism.
Semaglutide copies the GLP-1 mechanism. But two structural modifications change its pharmacology completely. These modifications solve the short half-life problem of native GLP-1.
Native GLP-1 is degraded at position 8. DPP-4 cleaves the Ala8-Glu9 bond. Semaglutide replaces Alanine at position 8 with alpha-aminoisobutyric acid (Aib8). DPP-4 cannot cleave this modified bond. Semaglutide stays active in circulation.
A second change, Arg34 to Lys34, has minimal effect on receptor potency. It mainly serves as the attachment point for the fatty acid side chain described below.
Semaglutide carries a C18 fatty acid side chain. This chain is attached at Lys26 via a γGlu-2xOEG (mini-PEG) linker. This modification enables semaglutide to bind reversibly to human serum albumin (HSA).
Molecular dynamics studies show the primary albumin binding site is the FA3-FA4 site on HSA. Albumin binding does two things. It prevents renal clearance. It also protects semaglutide from enzymatic degradation in circulation.
The result: semaglutide’s plasma half-life extends to approximately 165 hours. about one week. Compare that to native GLP-1 at 1-2 minutes and liraglutide at approximately 13 hours. Fatty acids from C12 to C20 were systematically screened. C18 di-acid with a γGlu-2xOEG linker showed the best albumin affinity and GLP-1R potency. Therefore, it was selected over other fatty acids. (Frontiers in Endocrinology, 2019)
Semaglutide forms more polar contacts with GLP-1R than native GLP-1. The specific hydrogen bonds and ionic interactions increase receptor affinity. The extended residency time of semaglutide at GLP-1R leads to more sustained receptor activation.
Semaglutide’s strong albumin binding may also influence receptor trafficking. Prolonged receptor engagement may shift the balance between G protein signalling and beta-arrestin recruitment. This is part of active research into biased agonism.
GLP-1R is not limited to one tissue. It is expressed across many organ systems. This wide GLP-1R tissue distribution explains why the GLP-1 mechanism produces effects beyond what a single-organ model would predict.
Pancreatic beta-cells: Highest GLP-1R expression. The cAMP-PKA-EPAC2 signalling cascade operates here in full. Glucose-stimulated insulin secretion (GSIS) is the primary output.
Pancreatic alpha-cells: GLP-1R activation suppresses glucagon release during high-glucose states. This glucagonostatic effect may work through direct receptor activation or via somatostatin from delta cells. (PMC, Revisiting GLP-1 Complexity, 2021)
Hypothalamus and brainstem: GLP-1R is expressed in the nucleus tractus solitarius (NTS) and arcuate nucleus (ARC). These regions process satiety signals. GLP-1 from intestinal L-cells also reaches CNS nuclei via vagal afferent pathways.
Cardiovascular tissue: GLP-1R is present in cardiac muscle and vascular endothelium. Research from 2024-2025 identifies anti-inflammatory and endothelial-protective actions through the GLP-1 signalling mechanism. Exact molecular pathways remain under active study.
Kidney: GLP-1R activation increases renal perfusion. Mechanistically, this is mediated by atrial natriuretic peptide (ANP) secretion. In tubular cells, PKA and PKC signalling provide additional effects. (Frontiers in Pharmacology, 2025)
Mesolimbic system: GLP-1 receptors are expressed in brain reward circuits, specifically mesolimbic and mesocortical pathways. This is an emerging area of GLP-1 receptor mechanism research as of 2024-2025. (PMC Frontiers Pharmacology)
Biased agonism is a key concept in advanced GLP-1 mechanism research. Different ligands can bind the same receptor. But they do not always produce the same intracellular output.
Some GLP-1R ligands prefer G protein coupling. They drive the cAMP-PKA-EPAC2 pathway strongly. Others favour beta-arrestin recruitment. Beta-arrestin triggers receptor internalisation and trafficking.
Cryo-EM structural studies show that biased agonists stabilise distinct GLP-1R conformations. The extracellular surface of GLP-1R acts as a molecular switch. The binding geometry of the ligand at the ECD determines which intracellular pathway is activated. (Nature, 2018; Endocrine Reviews, 2023)
GIP receptor (GIPR) couples selectively to Gαs. In pancreatic beta-cells, GLP-1R activates both Gαs and Gαq signalling pathways. This difference is pharmacologically important.
Tirzepatide is a GIP/GLP-1 dual agonist. It shows biased agonism at GLP-1R, preferential cAMP generation compared to balanced GLP-1 peptides. Combined with potent GIPR activation, this dual profile produces a distinct signalling output. Research shows the downstream signalling cascades of GLP-1R and GIPR overlap in some areas, such as adenylyl cyclase/cAMP activation, but diverge in others. (Frontiers in Endocrinology, 2024)
The GLP-1 mechanism starts with GLP-1 binding its class B G protein-coupled receptor (GLP-1R). This activates Gαs, raises intracellular cAMP via adenylyl cyclase, and activates PKA and EPAC2. These effectors close KATP channels, depolarise the cell membrane, trigger Ca2+ influx, and drive calcium-dependent cellular responses. The full GLP-1 signalling pathway is glucose-dependent; it requires sufficient intracellular ATP to complete.
Semaglutide has two key structural changes. First, Aib8 substitution at position 8 blocks DPP-4 degradation. Second, a C18 fatty diacid at Lys26 via a γGlu-2xOEG linker enables reversible albumin binding at the FA3-FA4 site on HSA. Together, these modifications extend the plasma half-life from 1-2 minutes (native GLP-1) to approximately 165 hours, enabling once-weekly dosing.
cAMP (cyclic AMP) is the primary second messenger in the GLP-1 mechanism. It is generated by adenylyl cyclase after Gαs activation. Elevated cAMP activates two effectors: PKA (which phosphorylates downstream targets and promotes gene expression via CREB) and EPAC2 (which lowers the threshold for KATP channel closure). A key finding: cAMP is also produced inside endosomes after receptor internalisation, a second wave of signalling.
GLP-1R has a wide tissue distribution. It is expressed in pancreatic beta- and alpha-cells, hypothalamic and brainstem nuclei (NTS, ARC), cardiovascular tissue, kidneys, lungs, gastrointestinal tract, and mesolimbic brain circuits. This widespread GLP-1R expression is why the GLP-1 mechanism produces effects across multiple organ systems, not just in the pancreas.
Biased agonism means different ligands bind GLP-1R but activate different intracellular pathways preferentially. G protein-biased agonists drive the cAMP-PKA-EPAC2 cascade. Beta-arrestin-biased ligands favour receptor internalisation. The extracellular domain of GLP-1R acts as a conformational trigger; the ligand’s binding geometry at the ECD determines which pathway dominates. Cryo-EM structural data from Nature (2018) and Endocrine Reviews (2023) confirm this mechanism.
Research Sources
PubMed / PMC · Nature (Signal Transduction and Targeted Therapy, 2024) · Frontiers in Endocrinology (2024) · Frontiers in Pharmacology (2025)
PNAS (2025) · American Journal of Physiology-Endocrinology and Metabolism · Scientific Reports (2017) · MDPI Pharmaceutics (2025)
Nutrition & Diabetes, Nature (2025) · Endocrine Reviews (2023) · The Discovery and Development of Liraglutide and Semaglutide, Frontiers in Endocrinology (2019)
No content in this article relates to human clinical use. All information is derived from molecular, cellular, and pharmacological research.
CagriSema is Novo Nordisk’s investigational fixed-dose combination of cagrilintide 2.4

Semaglutide is one of the most studied GLP-1 receptor agonists

Introduction Designing a precise GLP-1 titration schedule is one of

Semaglutide is one of the most studied peptides in modern